


本文首发药渡微信,作者: 杨柳青
1.序言
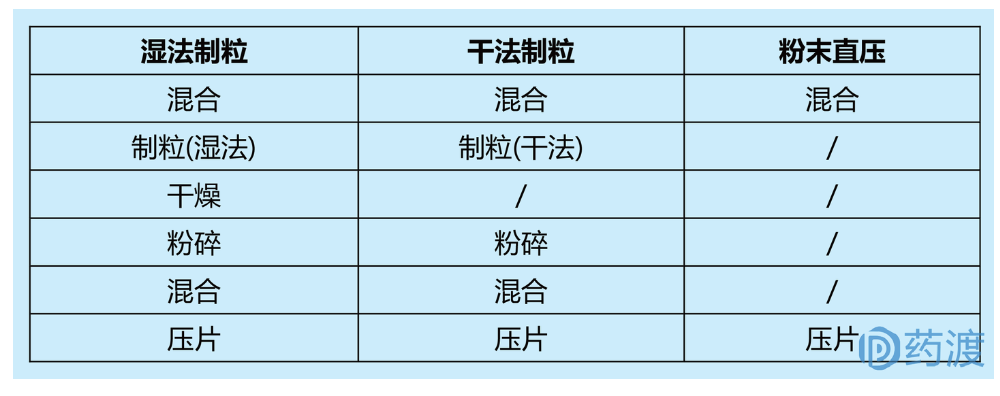
新药制剂开发应根据不同阶段选择合适的工艺,即“目的性”。如开发临床早期的制剂,应采用简单的工艺,便于新药能够快速进行临床概念性验证;当处于临床后期及商业生产的制剂,应充分考虑工艺的稳定性、可放大性等因素。固体口服制剂的主要单元操作包括制粒、干燥、粉碎、混合、压片(或灌装)等;其中根据制粒工艺,固体口服片剂常见三大工艺:湿法制粒、干法制粒和粉末直压工艺。下文将对新药固体口服制剂的常见工艺研究进行介绍。
2.制粒工艺
制粒主要目的是使原辅料粉末通过粘附形成较大颗粒(通常0.05-1.0mm),通过改善颗粒的粒径分布(PSD)、流动性、混合均一性等,便于后续灌装或压片过程。目前已有制粒技术如下图所示,其中最常用的制粒技术:干法制粒(碾压)和湿法制粒(高剪切制粒或流化床制粒)。
2.1 高剪切湿法制粒
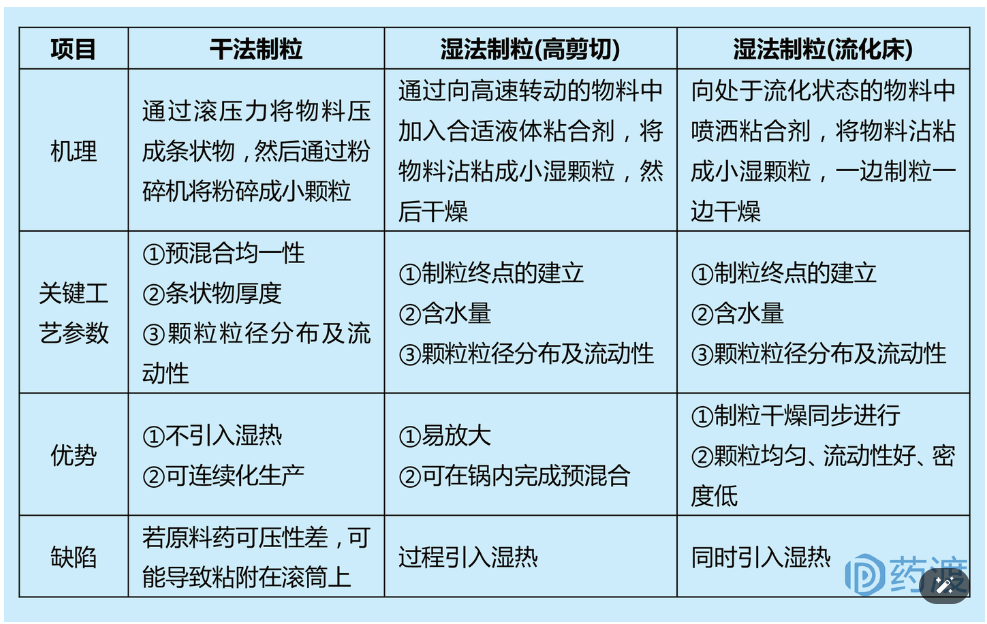
高剪切湿法制粒是目前制粒工艺中最常用的方法,与其他制粒技术相比,其工艺易于放大,颗粒易混合均一。为便于粒径生长,高剪切湿法制粒的载药量≤50%。高剪切湿法制粒机一般由锅体、喷嘴、搅拌桨和辅助切碎刀组成,依次完成物料预混合、加液制粒和补充制粒的过程。高剪切湿法制粒的许多工艺参数(如批量、粘合剂加入量、搅拌桨转速、制粒时间等)均会影响制得颗粒的质量。
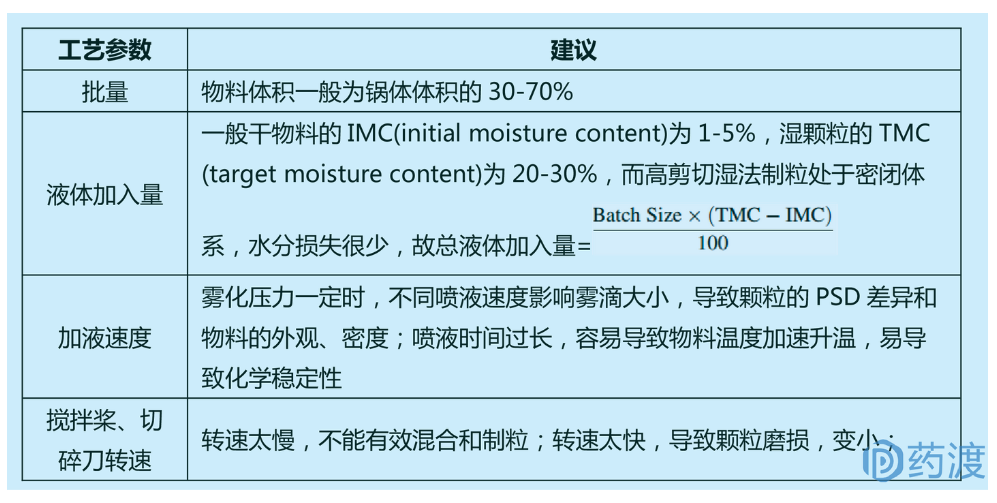
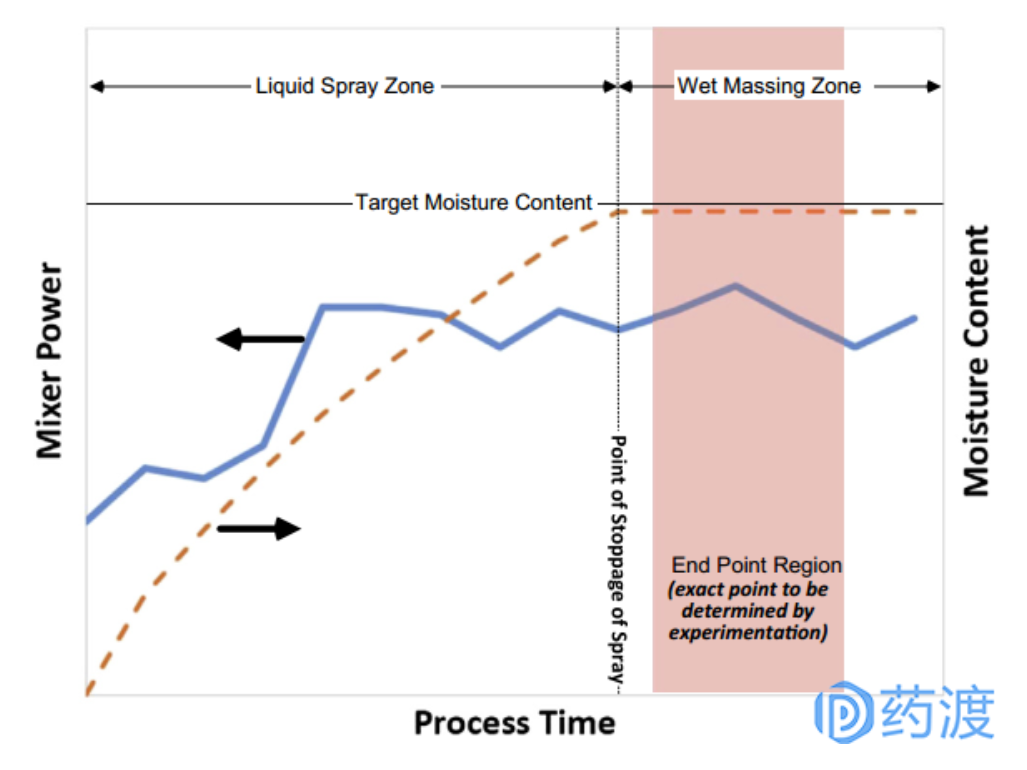
图:高剪切湿法制粒工艺过程
2.2 流化床制粒
流化床制粒是通过将粘合剂溶液喷洒到流化床粉末上生产颗粒的一种方法,其由物料混合、湿法制粒和干燥步骤组成。流化床中颗粒的运动轨迹、粘合剂的加入量和空气干燥能力对流化制粒影响较大。例如,较高的入口温度产生较细的颗粒,较低的温度产生较大的较强的颗粒。
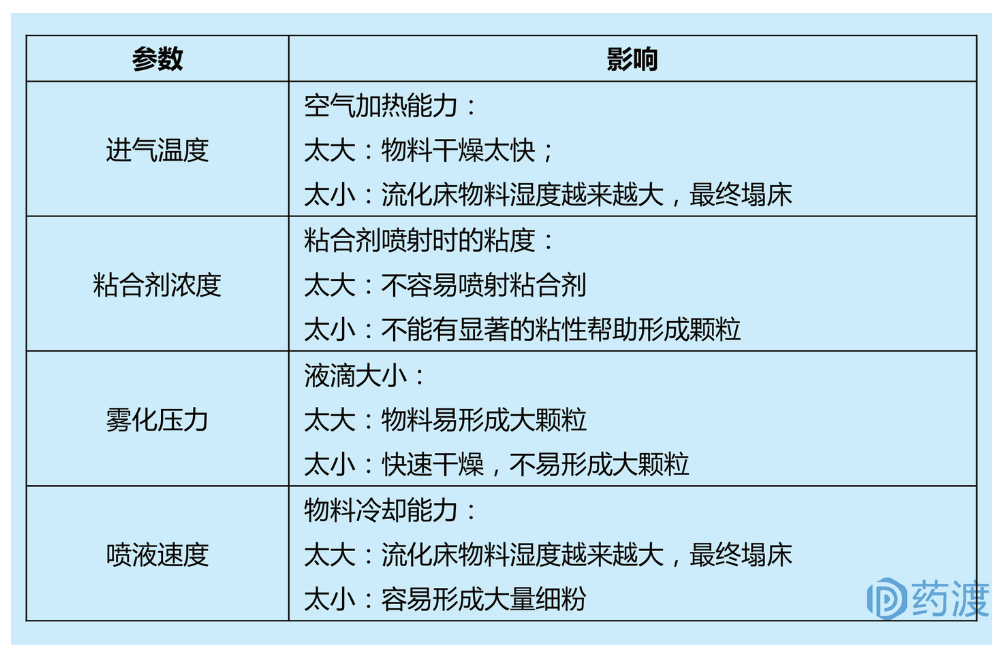
流化床制粒过程中,可以通过记录产品和排气温度、颗粒含水量等来监控工艺过程。
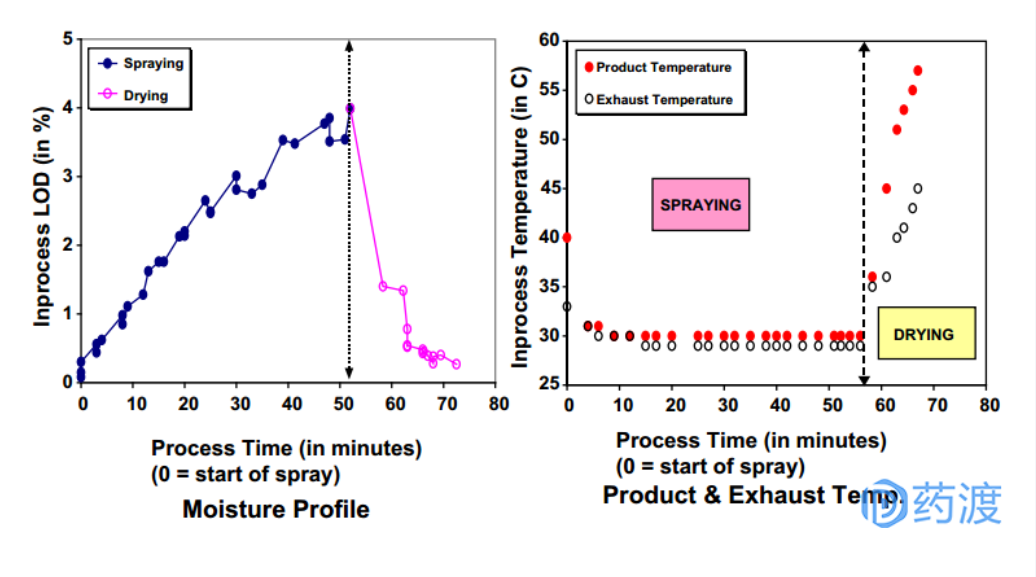
图:流化床湿法制粒工艺过程
2.3 干法制粒
干法制粒工艺过程中,混合均匀后的物料粉末在高压下压实、打碎、过筛后,形成一定粒径分布的颗粒。较湿法制粒,干法制粒的优势是工艺过程中不涉及水分和热量,适合湿热敏感物料制粒。同时,干法制粒占用空间较小,适合连续化生产。干法制粒不适用于可压性或二次可压性较差的物料;同时由于高压制粒,颗粒密度较高,可能减缓产品的溶出。
3.烘干工艺
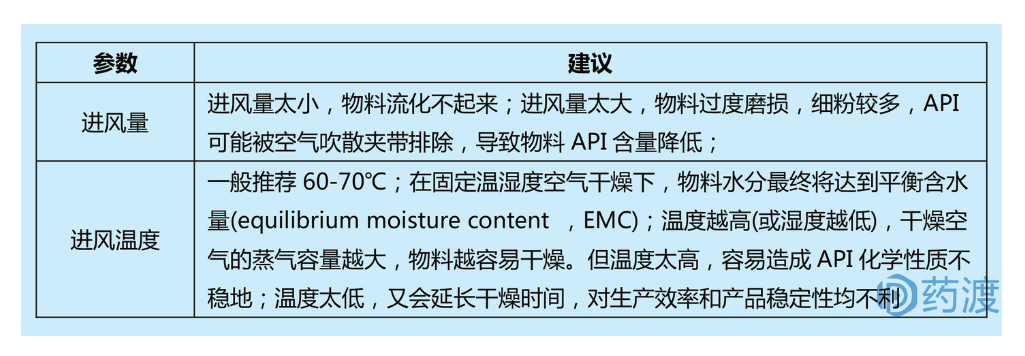
烘干工艺一般是湿法制粒(高剪切或流化床)后的必要步骤,常见干燥方式有鼓风烘箱、真空烘箱和流化床。其中流化床干燥工业应用较多,其通过热的进风空气将湿颗粒分散、流化,湿颗粒悬浮在流化空气中,热空气和湿颗粒之间的接触面积显著增大,能过在较短时间内实现干燥物料的目的。
4.粉碎工艺
经过干湿法制得的颗粒,粒径分布不均,流动性不好,容易导致混合不均一或片重差异较大;另外较大颗粒可能会减缓药物的溶出。通过颗粒的粉碎工艺,获得粒径分布较为均匀的颗粒,便于接下来工艺进行。常见的粉碎原理包括:挤压、撞击、摩擦、剪切。
干颗粒粉碎一般采用锥形研磨机,采用圆柱叶轮+圆孔锥形筛网,适合绝大多数干燥颗粒,具有低产热、少细粉。另外保持叶轮与筛网之间间隙很小,能够改善粒度分布均一性和粉碎效率。
5.混合工艺
干颗粒的混合工艺是保证最终药品含量均一性的关键步骤,混合机混合机制通常包括:对流、扩散、剪切。
混合前通常将助流剂、润滑剂或外部崩解剂加入粉碎好的干颗粒中,最常见的干颗粒混合机为翻滚式混合机,影响其混合效率的关键参数包括:物料批量、混合圈数(混合时间和速度)、加料顺序。
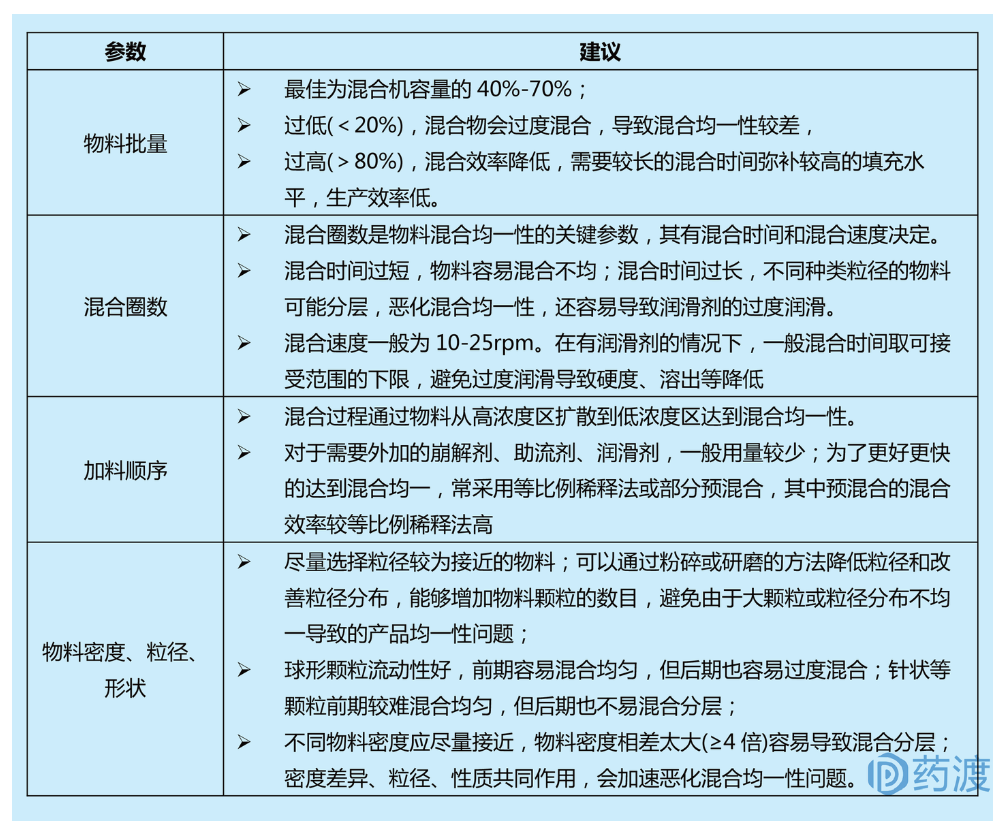
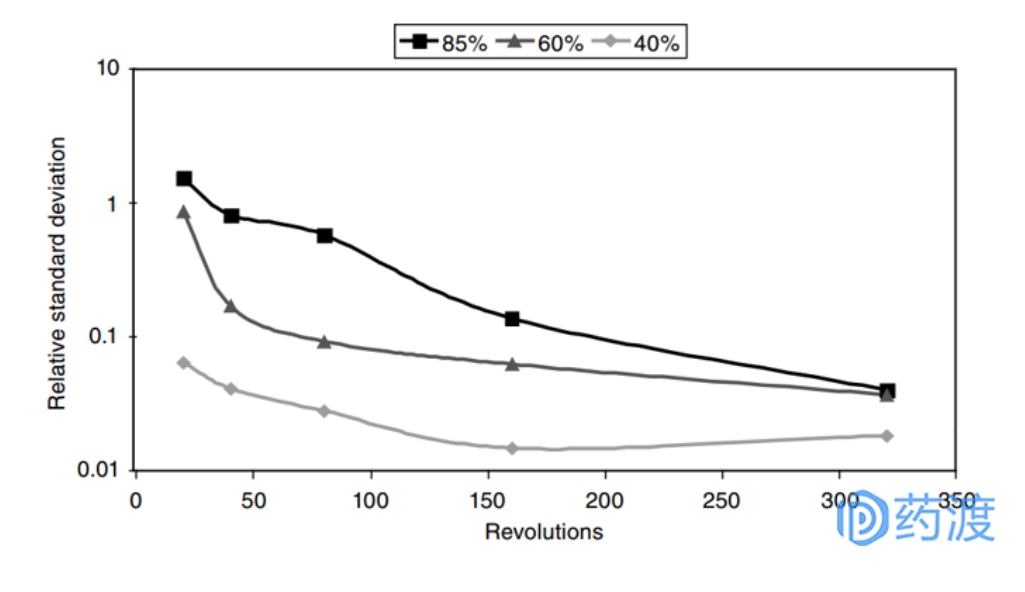
图:混合料筒不同装载量对混合效率的影响
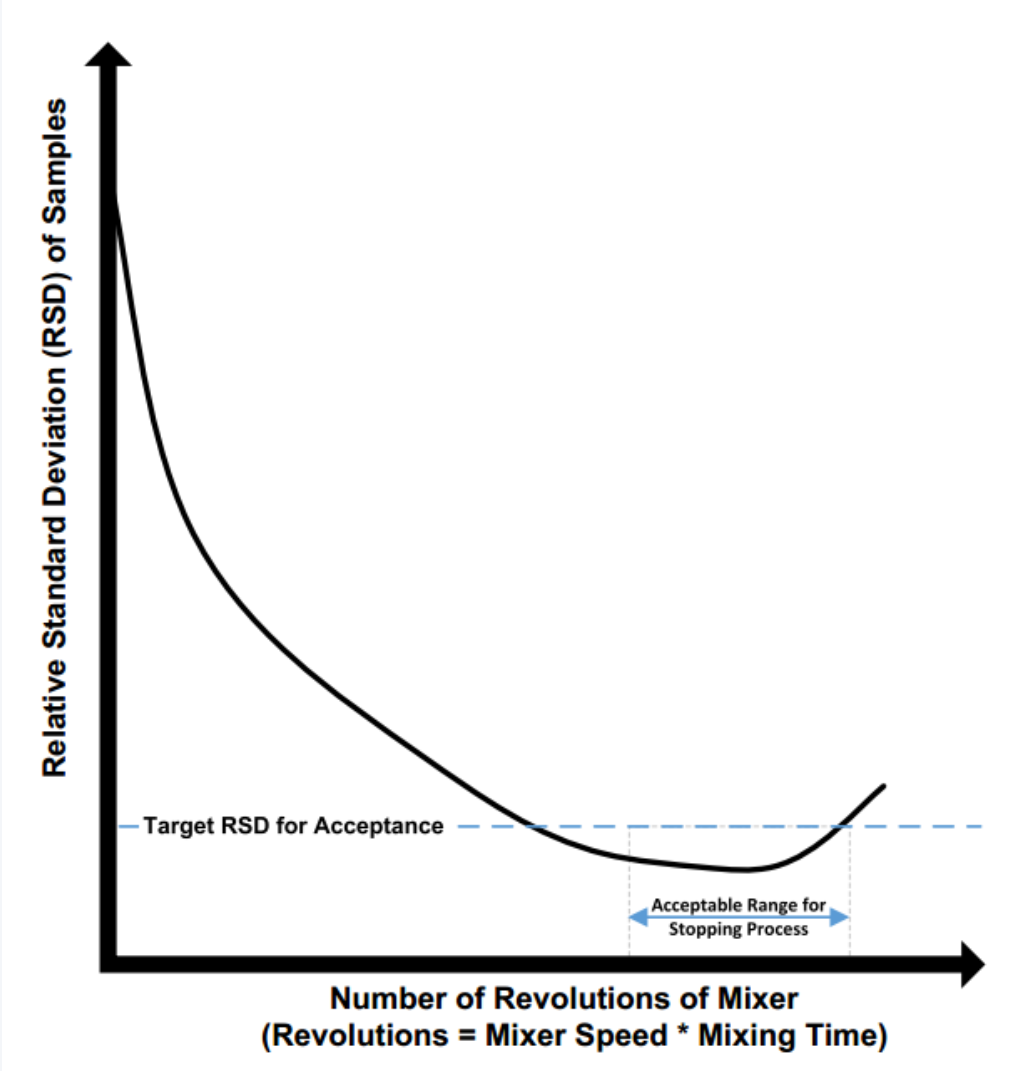
图:混合的工艺过程
评判混合均一性的方法主要采用取样检测混合料筒不同位置物料中API的含量是否一致。
6.压片工艺
压片工艺是将混合好的物料通过压片机成型的过程,其是临床给药的一种最终形式。压片过程:物料填充模具、调整填充重量、压实物料成片状、片剂从模具中分离。压片工艺的关键参数包括:主压力和转速。评价标准常为硬度和脆碎度。
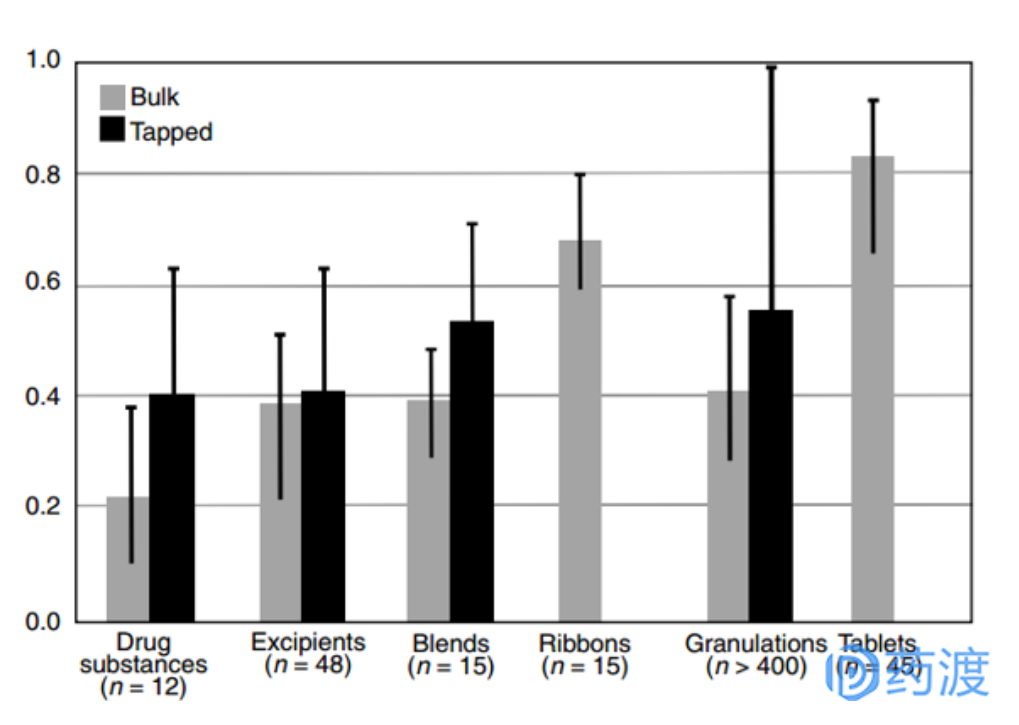
图:固体口服制剂中涉及物料的相对密度情况
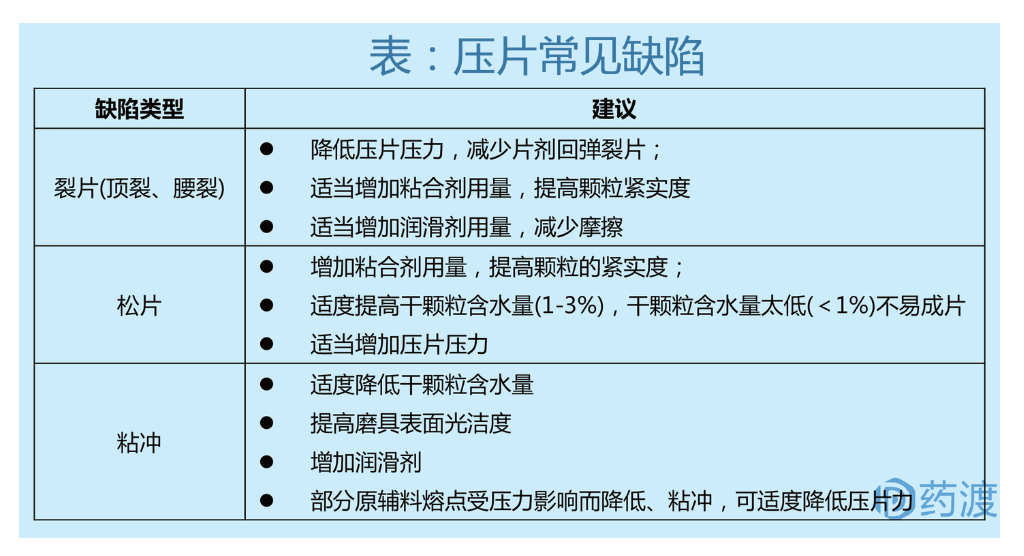
7.新药制剂工艺思考
7.1 制剂工艺的选择
固体口服制剂制备工艺最基础的单元操作是制粒。以片剂为例,制粒一般有三种选择:湿法制粒、干法制粒、粉末直压;制粒过程的选择取决于粉末混合的容易程度,即粉末流动性和混合均匀性。
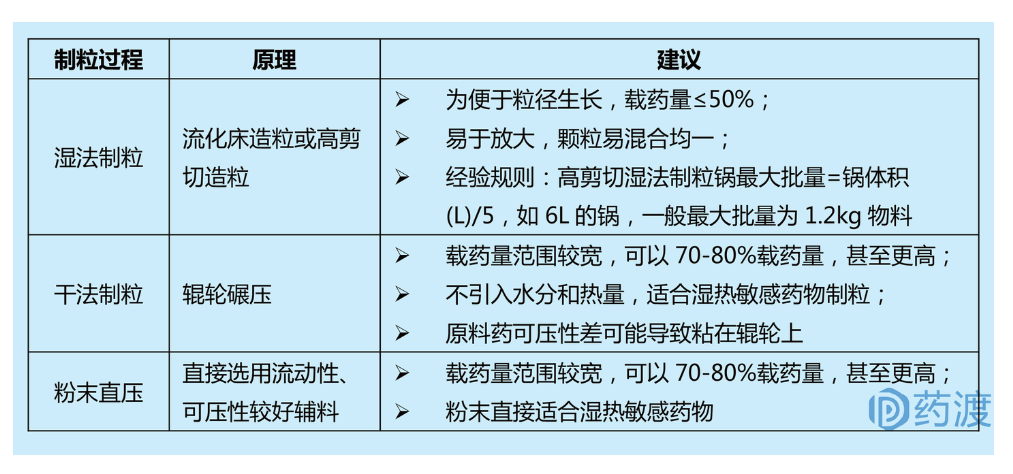
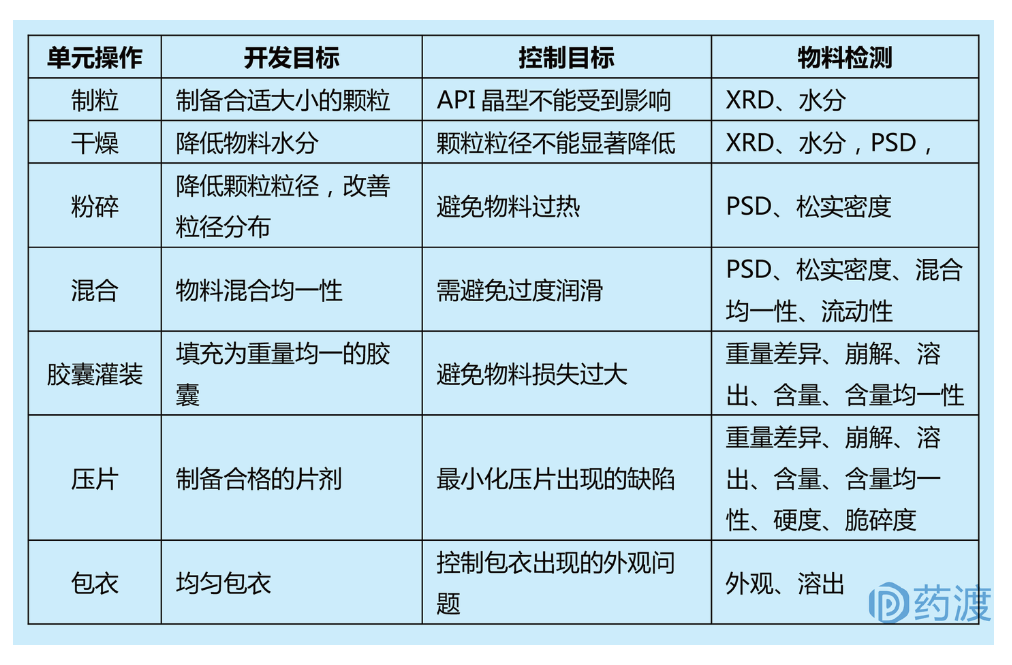
7.2 制剂工艺的开发目标
根据产品的TPP,需要对制剂工艺制定开发目标,以便达到TPP的要求。以典型的湿法制粒为例,介绍如何制定制剂工艺开发目标:
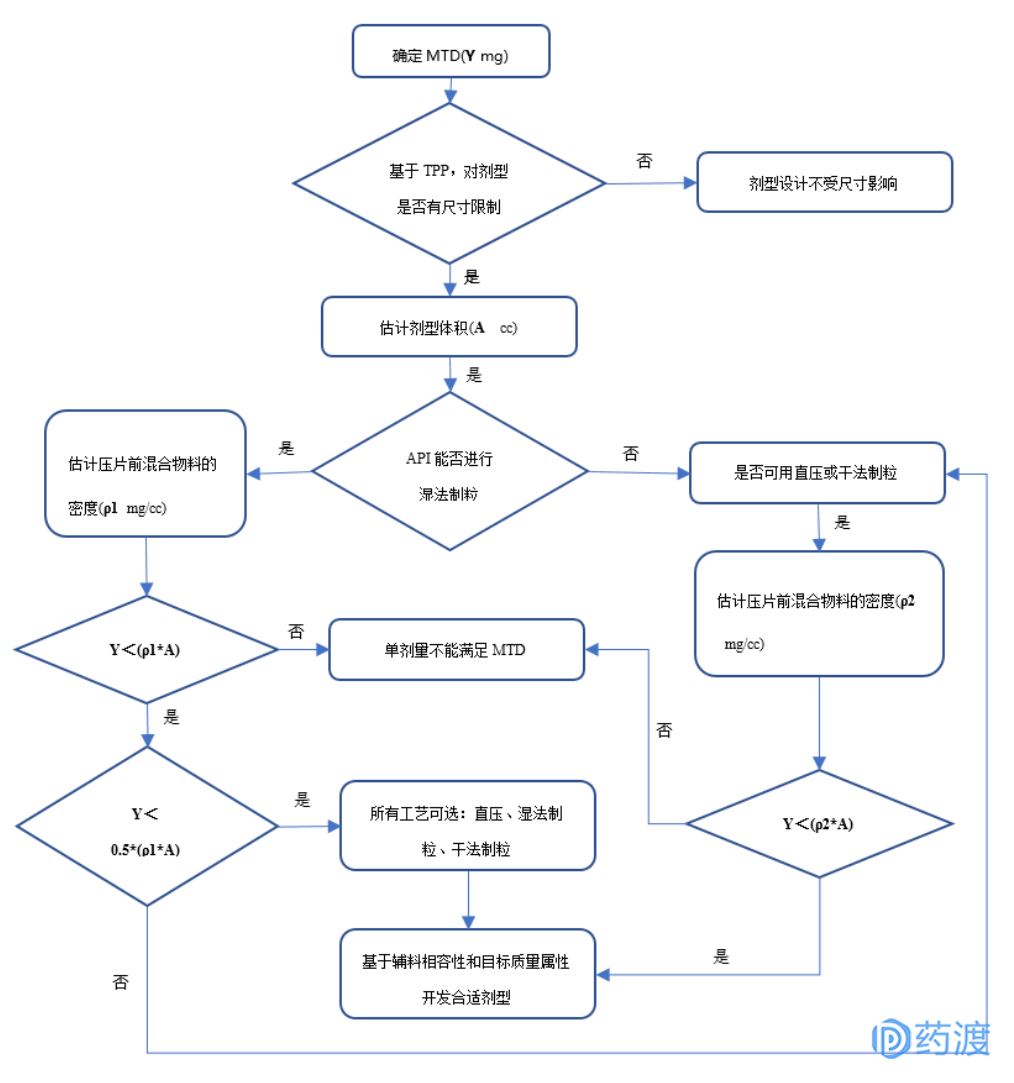
7.3 新药制剂开发决策
新药制剂的开发具有其特殊性,新化合物第一次开发成制剂,其很多性质还不甚了解,为了更有效率进行制剂开发,需要遵循一些基本原则和管理工具,如TPP等。在综合考虑了制剂处方工艺开发的基本原则,下表给出新药制剂开发的决策流程图:
通过对新药制剂进行合理、科学的设计,为新药加快进入临床试验提供有力保障。
项目研发前期准备
产品信息调研:在立项报告的基础上,对知识产权进行更新检索,确认不存在侵权的可能。然后针对处方工艺设计、质量标准建立等进行深入的文献检索,有条件的需获取进口质量标准或者
国内注册标准。
制订实验方案:制订详细的制剂实验方案和分析实验方案,列明项目参加人员、项目协助部门及相关人员的具体工作内容和介入时间点。
实验准备工作:
参比制剂的采购:首选已进口或本地化生产的原研产品;如果无法获得原研产品,可以采用质量优良的在发达国家上市的药品。如果上述国家产品已经进口中国,优先采用进口品;如果无法获得符合上述要求的参比制剂,则应在充分考虑立题合理性的前提下,采用多家国内上市的主流产品,进行深入的对比研究,所申报产品的质量应能达到其中最优产品的质量。
原料采购:可分别向几个厂家索取赠送小样,然后进行质量对比后,采购质量较好的原料药,需对方提供原料厂家资质、注册证、质量标准、检验报告、购销合同、发票等证明性文件。
色谱柱及对照品采购:在对原料质量标准以及查询到的制剂质量标准分析的基础上,拟定质量标准草案。向原料供应厂家充分了解产品的色谱条件后,再对色谱柱及对照品进行采购。对照品
选用标准:定量用对照品需采购USP、EP或者中检所的产品,定性用的对照品还可采购Sigma公司的产品。
辅料采购:根据国内辅料应用情况,对原研药的处方组成进行合理分析后索取不同厂家的赠送小样,经过预实验对比后,采购质量较好的辅料,生产部门已有的辅料可直接申请领用,新采购的辅料需对方提供辅料厂家资质、辅料注册证、质量标准、检验报告、购销合同、发票等证明性文件。辅料选用标准:首选药用级;无药用级,口服制剂可选用食用级。若也无食用级,考虑更换辅料。
包材的采购:在参比制剂购买或者获得准确的文献信息后,参考参比制剂的包装材料,结合公司情况,采购相应的包材,生产部门已有的包材可以直接申请领用,新采购的包材需对方提供包材厂家资质、包材注册证、质量标准、检验报告、购销合同、发票等证明性文件。
设备专用备件的订购:根据预实验样品进行设备备件的订购,如冲头、包装模具,这些部件的订购由研发部提供样品,设备科联系设计图纸,并经过主管领导批准后方可购买。
建立物料台帐,准备好仪器设备使用记录,做好预约计划。
处方工艺研究
原料及参比制剂的检验:小试用原料药按照质量标准进行关键项目的检验,应符合要求。对参比制剂进行全面的检测,包括尺寸、片重、硬度、崩解、有关物质、含量等,检测项目应不仅限于拟定的制剂质量标准,固体口服制剂应对参比制剂进行多种溶出曲线的测定。通过这些项目的检测,可以了解参比制剂的基本性能,为自研制剂的开发确立质量目标。
小试处方工艺摸索:
原辅料相容性试验:
如果通过前期的信息调研得知原料性质比较稳定,对辅料及保存条件没有太多要求时,若能查到原研药的处方组成,而拟定的辅料种类、厂家与原研药一致的情况下,可不做此试验;若在原研药处方的基础上,增加辅料种类,只需做增加的辅料相容性试验;若查不到处方组成,需做辅料相容性试验。具体做法:参照药物稳定性研究指导原则,将备选辅料分别与主药按一定比例混合,取一定量,在高温60度、强光、高湿(RH75%、RH92.5%)试验。分别于0天、(5天)、10天取样,重点考察性状、(含量)、有关物质等。同时用原料药和辅料分别做平行对照实验,以判别是原料药本身的变化还是辅料的影响。
如果对原料性质完全不了解,或通过信息调研得知原料性质不稳定,对辅料及保存条件有特殊要求时,即使查到原研药的处方组成,仍建议做辅料相容性试验,因为原辅料生产厂家不同,稳定性也不同,杂质种类也可能有差别。
处方筛选:通过上述原辅料相容性试验,对主药的稳定性有了基本的认识。先按照辅料的常规用量和常规工艺,以制剂基本性能(如口服固体制剂颗粒的可压性、流动性及药片的硬度、脆碎度、干燥失重等)为指标进行初步筛选。选出1 - 2个基本性能合格的处方样品,进行多种介质溶出曲线测定,与原研制剂进行比较,找出差距,调节辅料的用量或者浓度,使溶出曲线达到相似。对小试的关键工艺参数(如制粒转速、干燥温度等)进行考察筛选,初步确定制备工艺。
初步验证工艺:用拟定的处方工艺放大生产1 - 3批,样品规模片剂参考值为1000片/批。样品检验,检验标准为参照原研、国内首仿或国内主流产品、药典等拟订的本品质量标准草案,草案应不低于被仿标准。产品合格,并与参比制剂质量一致,则确定处方工艺;产品不合格,则重新进行处方工艺筛选。
影响因素试验:参照药物稳定性研究指导原则。用上述初步验证制备的样品,和原研产品对比进行影响因素研究。如果产品与原研药稳定性相似,则认为小试处方工艺基本可行,否则重新进行处方工艺筛选。建议此时同时放置裸片和包装后的样品,以降低包材不相容的风险。
中试生产及清洁预验证
中试批量:根据法规要求和公司现状拟定中试批量。
中试生产:在车间生产三批中试产品(不包括预试批)。
质量研究
质量标准制定:根据药典和相关法规,制定产品的质量标准,包括外观、含量、溶出度等指标。
检测方法学验证:对检测方法进行精密度、准确度、线性等方面的验证,确保检测结果的可靠性。
稳定性研究
对制剂进行加速和长期稳定性研究,评估其在不同条件下的稳定性,包括外观、溶出度、含量等指标的变化。
工艺验证及清洁验证
工艺验证:对生产工艺进行验证,确保工艺的稳定性和可靠性,能够生产出符合质量标准的产品。
清洁验证:对生产设备和生产环境进行清洁验证,确保清洁效果符合要求,避免交叉污染。
参考文献:
1、How to Develop Robust Solid Oral Dosage Forms
2、Pharmaceutical-Dosage-Forms-Tablets
3、FDA